
PEA: a Potent Buffer Enhancing Molecule
Palmitoylethanolamide (PEA): The Endogenous Compound That Helps Your Body Absorb Stress Without Breaking

The Endogenous Molecule That Stops Inflammation From Spiraling
Most people think stress damages them directly.
That’s incomplete.
Stress triggers counter-forces.
When tissue is injured, when LPS (lipopolysaccharide) hits TLR4, when mast cells activate, when microglia shift into inflammatory mode, your body does not passively accept damage.
It releases buffer mediators.
One of them is Palmitoylethanolamide (PEA).
PEA is not a foreign molecule/supplement.
It is an on-demand lipid mediator your cells synthesize when under stress.
Not as a stimulant, but rather as a protective brake.
What PEA Actually Is
PEA is an N-acylethanolamine (NAE).
Specifically:
Palmitic acid + ethanolamine → N-hexadecanoylethanolamine.
It is produced from membrane phosphatidylethanolamine via:
Ca²⁺-dependent N-acyltransferase (NAT)
NAPE-PLD (phospholipase D)
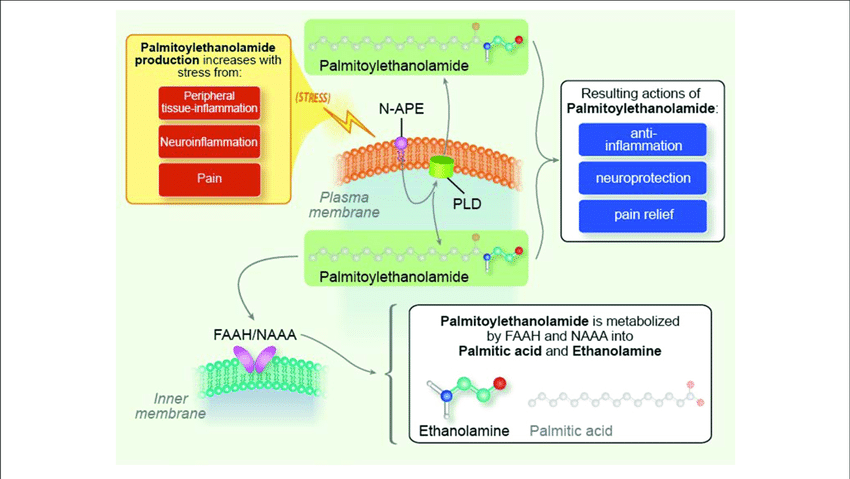
Translation:
When cells are stressed, they convert membrane lipids into PEA.
It is synthesized in:
Neurons
Glia
Immune cells
Gut tissue
It is not stored.
It is made when needed.
That’s important.
PEA is a compensation/buffering molecule.
The First Layer: Mast Cell Stabilization (ALIA)
When stress activates mast cells, they release:
Histamine
TNF-α
Proteases
Cytokines (promotes inflammation)
That cascade increases:
Vascular permeability
Neuroinflammation
Peripheral sensitization (pain sensitivity)
PEA downregulates mast-cell degranulation.
This is called:
Autacoid Local Injury Antagonism (ALIA).
Translation:
PEA limits how far the local inflammatory reaction spreads.
Same insult.
Smaller inflammatory radius.
That’s buffer function.
The Second Layer: PPAR-α (Genomic Anti-Inflammatory Hub)
PEA binds and activates PPAR-α.
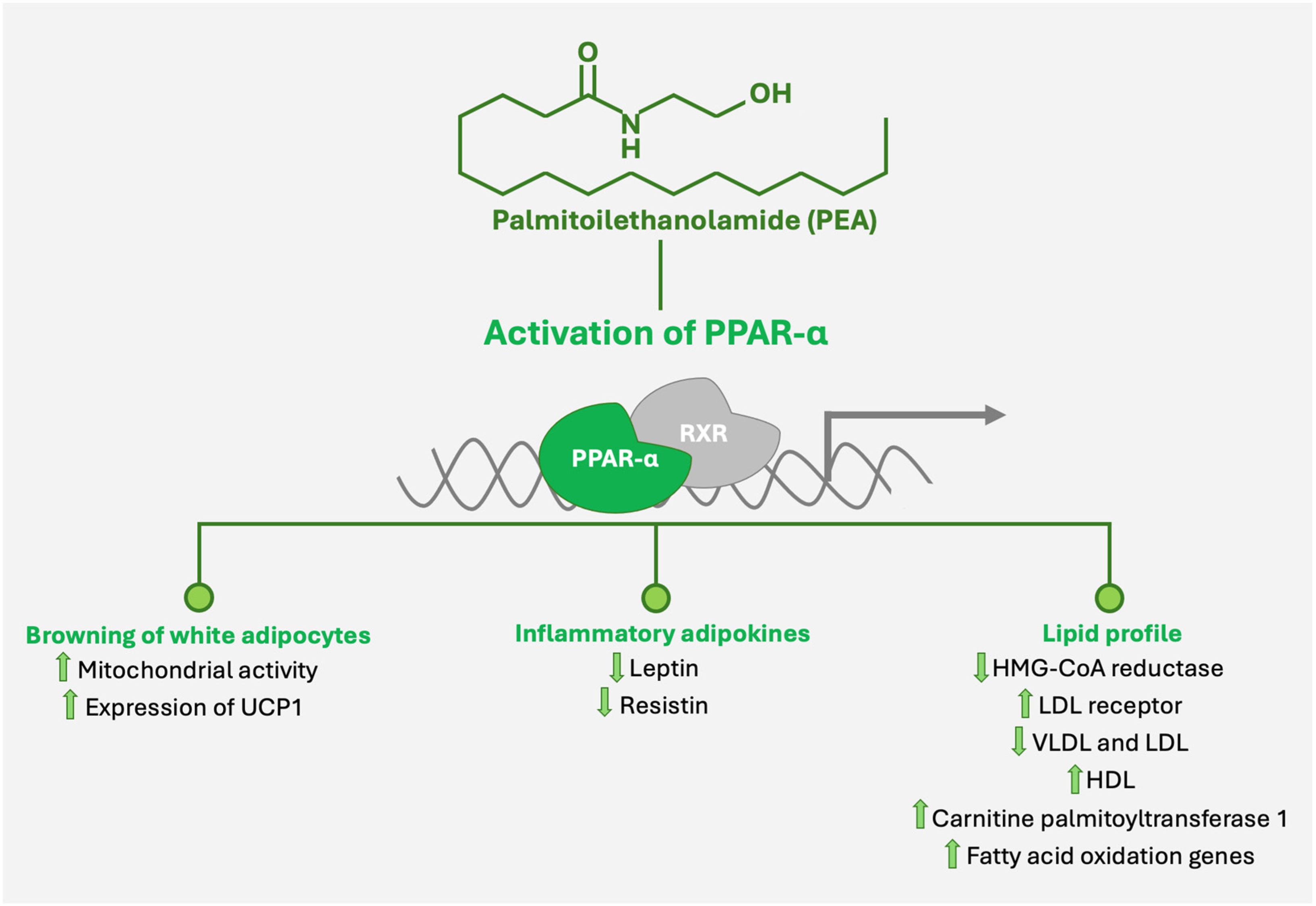
PPAR-α suppresses inflammatory pathways/markers like:
NF-κB transcription
TNF-α
IL-1β
IL-6
COX-2
iNOS
This shifts cells away from inflammatory transcriptional programs.
Not by blocking the immune system.
By changing gene expression.
It also improves:
Mitochondrial function
Fatty acid oxidation
Oxidative stress control
Stress normally activates NF-κB.
PEA limits how long it stays active.
That prevents inflammation from spiraling out of control and caps how long it stays active.
The Third Layer: Microglia and Neuroinflammation
Under LPS or metabolic stress, microglia shift toward an M1 phenotype.
M1 → pro-inflammatory → neurotoxic.
PEA shifts microglia toward M2:
Pro-resolving.
Phagocytic.
Cleaner of debris.
It also:
Blunts intracellular calcium rise (excitatory)
Reduces network hyperexcitability
Modulates CB2-linked immune tone
This is where PEA differs from agmatine.
Agmatine mainly modulates excitatory tone and NMDA/glutamate-related stress buffering.
PEA primarily modulates:
Neuroimmune activation and glial state.
Agmatine calms the signal.
PEA limits the inflammatory cascade.
Together?
You dampen excitotoxic stress and inflammatory amplification.
That’s synergy.
The Gut: Where This Gets Interesting
PEA is synthesized in the intestinal mucosa.
In colitis (inflammation or swelling of the colon) and ischemia models, it:
Reduces mucosal damage
Reduces TNF-α and IL-1β
Decreases NF-κB activation
Improves epithelial tight junction integrity
Translation:
It strengthens the intestinal barrier.
Less endotoxin translocation.
Less systemic inflammatory signaling.
And here’s the paradox:
High-fat diets can lower intestinal PEA levels in the medium term… despite high palmitate intake.
So more substrate does not automatically mean more PEA.
Endocannabinoid “Entourage” Layer
PEA does not directly activate CB1 or CB2 strongly.
But it:
Competes for FAAH/NAAA degradation
Slows breakdown of AEA (anandamide)
Enhances local endocannabinoid tone
This indirectly:
Reduces pain signaling.
Improves mood stability.
Modulates immune output.
Neurosteroids and Allopregnanolone
Several animal models show that PEA increases brain allopregnanolone levels by activating the PPARa pathway.
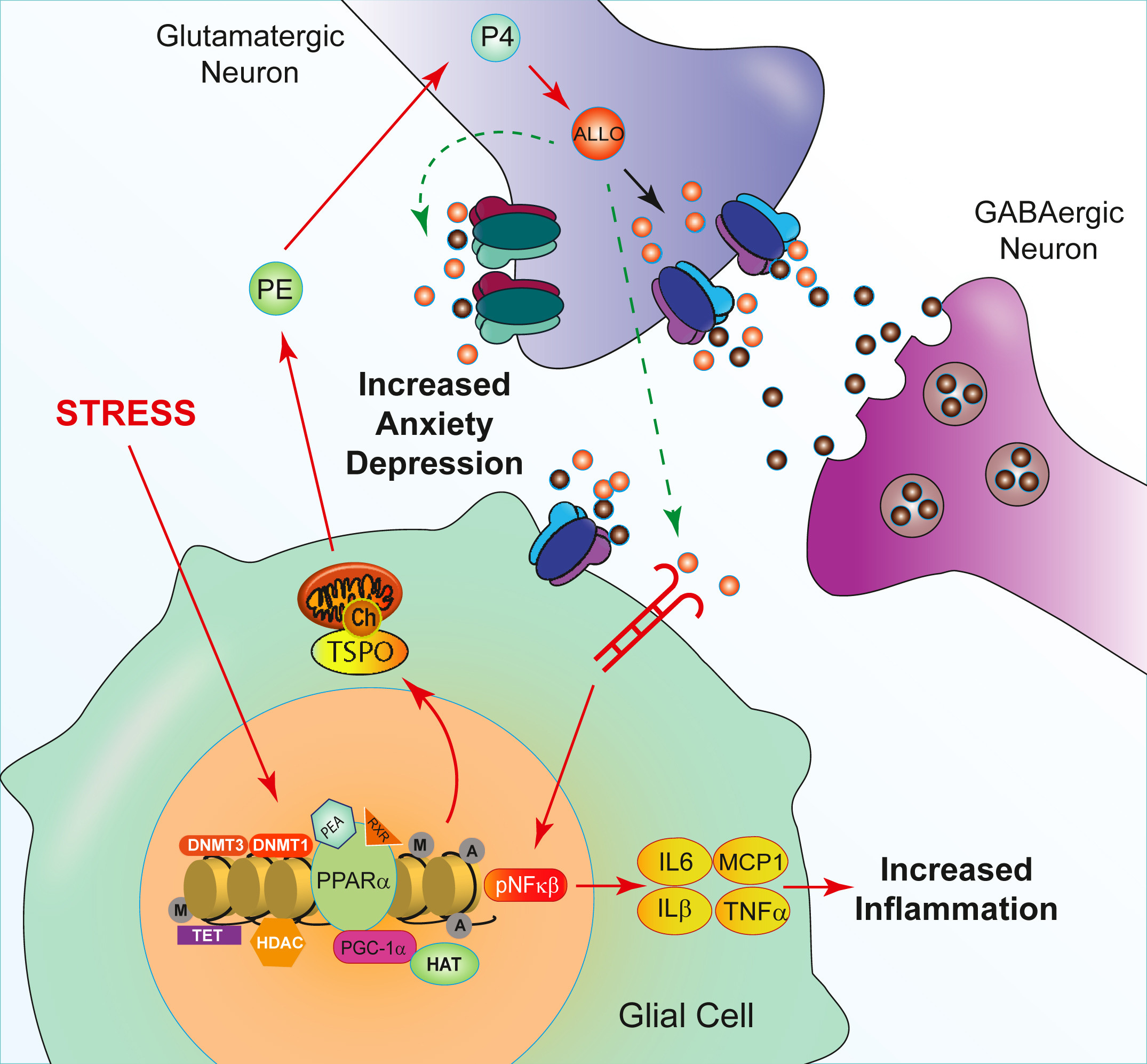
Allopregnanolone is a positive modulator of GABA-A.
Translation:
PEA reduces inflammation →
Glial state improves →
Neurosteroid production increases →
GABAergic tone rises.
Less glutamate dominance.
Less stress amplification.
PEA stabilizes inflammatory signaling and enhances neurosteroid buffering.
This helps prevent the anxiety-inducing effects of inflammation.
PEA, Finasteride, and Neurosteroid Compensation
Here’s where things get more nuanced.
As mentioned above, PEA actively stimulates neurosteroid synthesis.
In glial cells, PEA:
→ Activates PPARα
→ Increases StAR and P450scc expression
→ Increases de novo allopregnanolone synthesis
Allopregnanolone is not trivial.
It is one of the most powerful endogenous positive GABA receptor modulators in the brain.
Meaning, it makes the GABA receptor work better.
Higher allopregnanolone:
Lowers anxiety
Reduces hyperexcitability
Buffers stress signaling
Improves mood resilience
In rodent models, PEA increases spinal and brain allopregnanolone levels.
And here’s the critical part:
When 5-alpha-reductase is blocked (with finasteride),
PEA’s behavioral and analgesic (pain killing) benefits are significantly reduced.
That tells you something important:
PEA’s central effects partially depend on intact 5α-reductase activity.
Why This Matters for Post-Finasteride Syndrome (PFS)
PFS patients show significantly reduced neurosteroids in CSF (cerebrospinal fluid)
…including:
Progesterone metabolites
DHP
Allopregnanolone
This neurosteroid collapse is strongly associated with:
Anxiety
Depression
Sexual dysfunction
Cognitive issues
Post partum depression
Mechanistically, PEA makes sense here:
PEA → PPARα activation → increased neurosteroidogenesis → increased allopregnanolone
But:
If 5α-reductase activity is impaired, PEA’s ability to raise downstream neurosteroids will be partially constrained.
That’s the paradox.
PEA is a neurosteroidogenic enhancer.
But it relies on the same enzyme finasteride inhibits.
So at present:
There are no controlled human trials using PEA specifically in diagnosed PFS.
What we have is:
PEA → allopregnanolone in glial models
Finasteride blocking PEA’s neurosteroid-mediated effects in rodents
Neurosteroid deficits documented in PFS patients
That makes PEA a mechanistically compelling candidate.
But still theoretical in confirmed PFS cohorts.
That distinction matters.
Now let’s integrate the gut axis cleanly. (can’t skip gut on my watch!)
PEA and the Gut–Brain Axis: The Serotonin Link
Another layer most people miss:
PEA modulates tryptophan metabolism.
In high-fat diet models:
Obesity reduced colonic serotonin (5-HT)
enhanced risk of constipation
Increased 5-HIAA
enhanced serotonin turnover, meaning less colonic serotonin available
Shifted tryptophan toward the kynurenine pathway (↑ IDO, ↑ KYN)
“Tryptophan steal” so to speak, meaning less tryptophan available for muscle growth, transit time and brain serotonin synthesis.
The metabolites of the kynurenine pathway, especially quinolinic acid, are highly excitatory, which contributes to anxiety and neurological damage.
Translation:
More inflammation →
More IDO activation →
Less serotonin →
More kynurenine metabolites →
More systemic and neuroinflammatory signaling.
PEA reversed this pattern.
It:
Increased colonic 5-HT
Reduced 5-HT turnover
Reduced IDO expression (blunted kynurenine pathway activation)
Reduced (excess) kynurenine levels
This is major.
Because IDO is upregulated by inflammatory pathways like:
TNF-α
IL-1β
TLR4 activation
Which means:
Inflammation → tryptophan shunted into KYN → mood and metabolic disturbance.
PEA dampens the inflammatory trigger.
Tryptophan stays in the serotonin pathway.
Mood stability improves.
But there’s more.
PEA reshaped the microbiota.
It:
Reduced LPS and hydrogen sulfide (inflammatory in excess)-producing taxa (Bilophila, Desulfovibrio)
Increased butyrate-linked taxa (Oscillospiraceae, Bifidobacterium)
Increased Turicibacter sanguinis (a serotonin-sensing microbe)
It also increased:
GPR43 (butyrate receptor)
MCT1 (butyrate transporter)
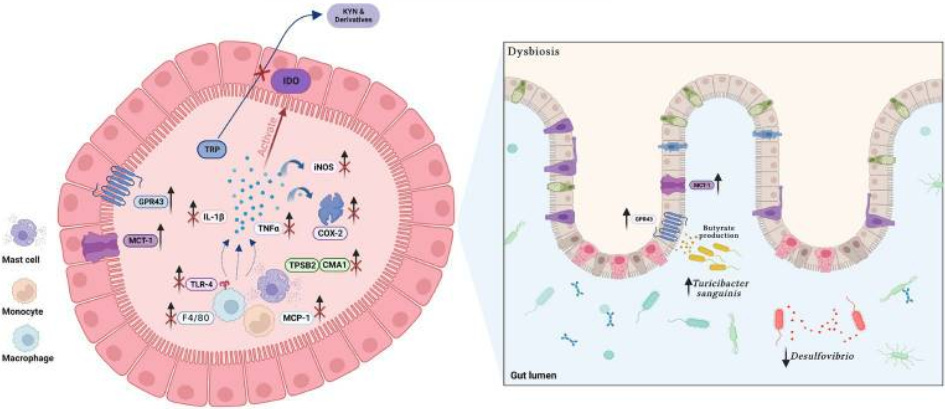
Translation:
PEA doesn’t just reduce inflammation.
It increases the gut’s responsiveness to SCFAs. In other words, never supplement butyrate without PEA. It’s like training hard in the gym without adequate testosterone… a waste of time.
So:
Less endotoxin.
Better barrier.
Better TRP handling.
Better serotonin tone.
Again:
That’s buffer expansion.
The Big Picture
PEA is not a “pain supplement.”
It is an endogenous anti-inflammatory amplifier released when tissues detect stress.
Its roles span:
Mast-cell regulation
Microglial phenotype shifting
NF-κB modulation
Mitochondrial stabilization
Barrier integrity
Endocannabinoid tone
Neurosteroid production
It reduces the net damage of a stressor without suppressing necessary immune function.
That’s the definition of a buffer molecule.
And here’s the key insight:
If chronic stress depletes or dysregulates PEA production,
inflammation lingers longer than it should.
That is where supplementation can become strategic.
What We’ll Cover Next
Understanding what PEA does is only half the story.
The real question is how to use it.
Because the effects of PEA depend heavily on dose, timing, and context. It can be used to lower background inflammation during the day, increase pain tolerance before training, or support nervous system down-regulation in the evening.
In the rest of this article, we’ll cover:
How much PEA to take
Which form and brand is best
When to take it depending on your goal
How to stack it with synergistic compounds
Who should be cautious with it
And how to increase your body’s own PEA production naturally
If you want the practical protocol, that’s what we’ll go through next.
